































製品情報
製品情報医療用医薬品を適正にご使用いただくための情報を提供しています。
セミナー・講演会情報

動画ライブラリ

疾患・領域情報
疾患・領域情報疾患ならびに領域において、お役立ていただける専門性の高い情報を提供しています。骨粗鬆症DICリウマチ
医療現場最前線
クローズアップ
医療現場最前線医療関係者が「知りたい」「聞きたい」現場の声をお届けしています。
医療情報

旭化成ファーマの取り組み
旭化成ファーマの
取り組み旭化成ファーマの取り組みに関する情報を提供しています。

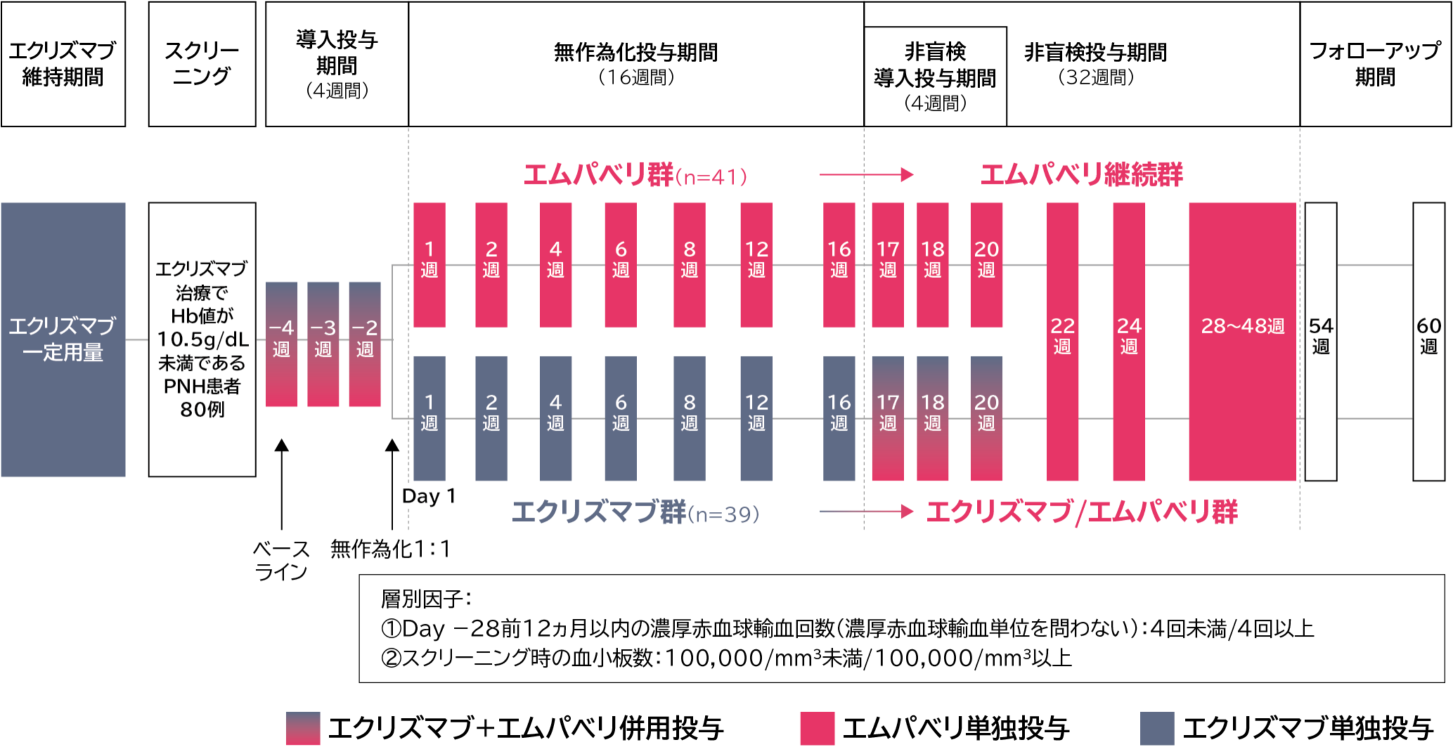
国際共同第Ⅲ相試験[PEGASUS試験]
試験概要
| 目的 | エクリズマブ治療でHb値が10.5g/dL未満であるPNH患者に対して、エムパベリを投与したときの有効性及び安全性を、エクリズマブ継続投与と比較する。 |
|---|---|
| 試験デザイン | 前向き、無作為化、多施設共同、非盲検、実薬対照比較試験 |
| 対象 | 高感度フローサイトメトリーを用いてPNHと診断された18歳以上の男女で、スクリーニング来院前3ヵ月間にわたる一定用量のエクリズマブ治療でHb値が10.5g/dL未満であった患者80例(日本人10例を含む) |
| 方法 |
本試験の投与期間は、導入投与期間[4週間:−4週〜Day 1(治験薬投与前)]、無作為化投与期間(16週間:Day
1~16週)及び非盲検投与期間(32週間)とした。
|
| 主な選択基準 |
|
| 主な除外基準 |
|
| 有効性評価項目 |
〔主要評価項目〕
〔重要な副次評価項目〕
〔その他の副次評価項目〕
|
| 安全性評価項目 |
|
| 薬力学評価項目 |
|
| 解析計画 |
主要評価項目の解析
主要評価項目は、Intent-to-treat(ITT)集団※1を対象に無作為化投与期間の16週時点のHb値のベースラインからの変化量とした。被験者が無作為化投与期間中に輸血を受けた場合又は試験中止となった場合は、輸血まで又は試験中止時までのHb値を解析対象とした。主要評価項目の解析は、16週時点におけるエムパベリ及びエクリズマブのHb値のベースラインからの変化量を求め、群間比較を反復測定による混合モデル(MMRM)を用いて評価した。また、両側95%信頼区間(CI)及びp値を算出した。主要評価項目の結果への輸血による影響を回避するため、初回輸血後のすべての値をデータから除外し、MMRMを用いて、ベースライン、層別因子、投与、来院及び投与群と来院の交互作用で調整したデータを解析した。主要評価項目に対して、輸血で打ち切られた対照群のデータに基づくパターン補完及びTipping Point法に基づく補完の2種類の感度分析を実施した。
重要な副次評価項目の解析
重要な副次評価項目の解析は、非劣性検定に基づき評価した。階層的検定手順に従い重要な副次評価項目の検定を行った。非劣性が検証されない変数があった場合、その後のすべての検定を実施しないこととした。すべての重要な副次評価項目で非劣性が示された後に、輸血回避割合、無作為化投与期間の16週時点の網赤血球数のベースラインからの変化量、LDH値のベースラインからの変化量及びFACIT-Fatigueスコアのベースラインからの変化量の有意性を評価した。無作為化投与期間の16週時点の網赤血球数、LDH値及びFACIT-Fatigueスコアのベースラインからの変化量は、各評価項目のベースラインを共変量とし、ITT集団及び改変ITT(mITT)集団※2を対象としたこと以外は、主要評価項目の解析と同じ方法を用いて評価した。輸血回避割合については投与群別に示し、層化Cochran-Mantel-Haenszelのχ2検定を用いて投与群間比較を実施した。重要な副次評価項目の非劣性の達成要件は、輸血回避割合では群間差の95%CIの下限値が非劣性マージンの-20%より大きい場合、網赤血球数では群間差の95%CIの上限値が非劣性マージンの10×109/Lより小さい場合、LDH値では群間差の95%CIの上限値が非劣性マージンの20U/Lより小さい場合、FACIT-Fatigueスコアでは群間差の95%CIの下限値が非劣性マージンの-3より大きい場合とした。
その他の副次評価項目の解析
その他の副次評価項目の解析では、ITT集団を用いて48週時点までの実測値及びベースラインからの変化量を、無作為化投与期間及び治験期間全体の各来院ごとに投与群別に要約した。16週までの無作為化投与期間における評価項目では、主要評価項目及び重要な副次評価項目と同様に、輸血によるデータの打ち切りを実施した。
サブグループ解析
サブグループ解析として、Day –28前12ヵ月以内の濃厚赤血球輸血回数(4回未満、4回以上)部分集団の主要評価項目及び重要な副次評価項目の結果を要約し、解析した。また、日本人集団における主要評価項目、重要な副次評価項目及びその他の副次評価項目について、実測値及びベースラインからの変化量(輸⾎によるデータの打ち切りあり)に関する記述統計量を示した。
安全性の解析
安全性の解析は、安全性解析対象集団を対象に実施した。安全性解析対象集団は、無作為化され、治験薬投与を1回以上受けたすべての被験者を含む集団とした。全集団に加え、日本人集団についてのサブグループ解析も行った。
薬力学の解析
薬力学の解析は、ITT集団のうち治験薬投与後に評価可能な薬力学測定値を1つ以上有するすべての被験者を含む集団(PD集団)を対象に実施した。PNHタイプⅡ+Ⅲ赤血球、C3オプソニン化PNHタイプⅡ+Ⅲ赤血球の実測値、無作為化投与期間の16週時点のベースラインからの変化量の記述統計量を各来院ごとに投与群別に示した。 |
有効性[無作為化投与期間(16週間)] 全集団
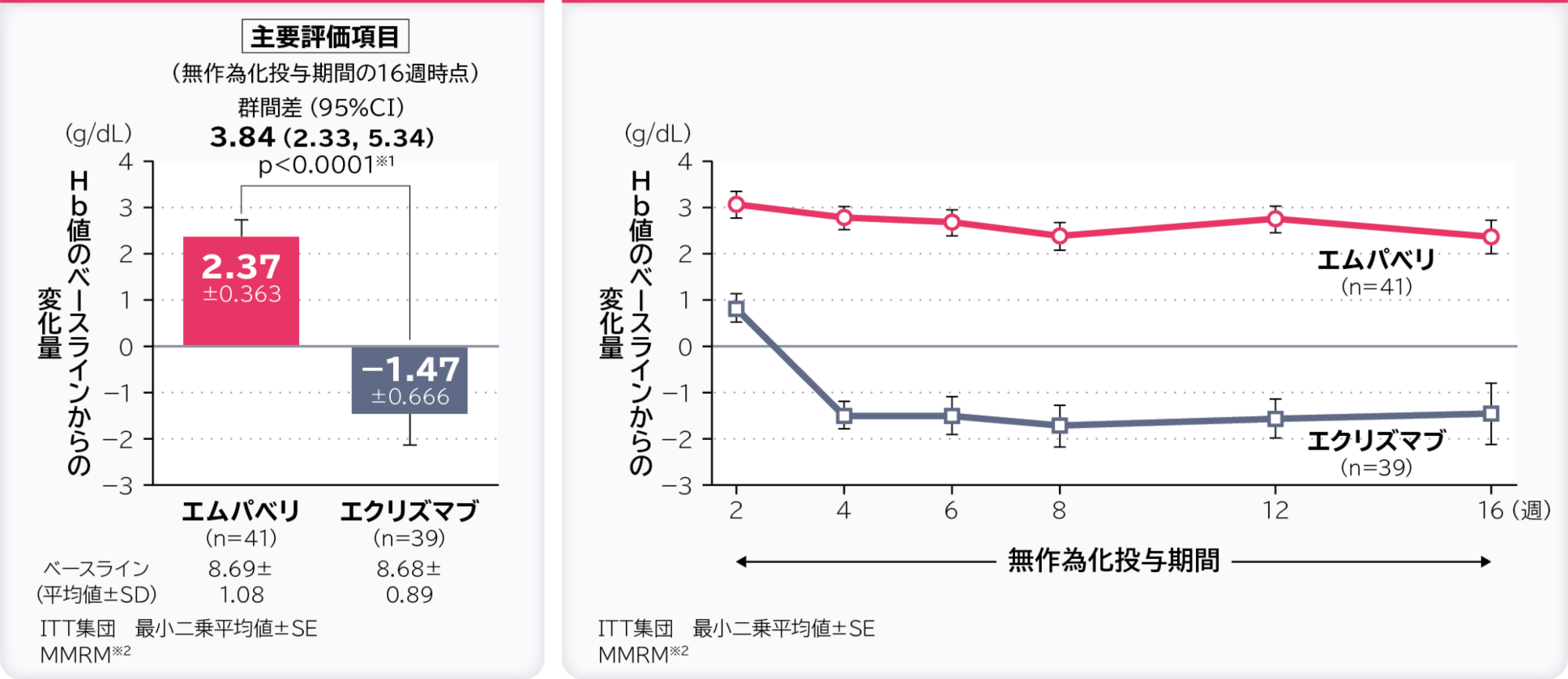
1. 無作為化投与期間の16週時点のHb値のベースラインからの変化量〔主要評価項目〕 検証的解析結果
無作為化投与期間の16週時点のHb値のベースラインからの変化量の調整平均(最⼩⼆乗平均値)はエムパベリ群2.37g/dL、エクリズマブ群-1.47g/dL、群間差は3.84g/dL(95%CI:2.33, 5.34)であり、エムパベリ群ではエクリズマブ群に比べてHb値の変化量に有意差が認められ、エムパベリ群のエクリズマブ群に対する優越性が検証されました。
- ※1:有意水準α=0.05
- ※2:ベースライン、層別因子、投与、来院及び投与群と来院の交互作用で調整したデータを解析した。
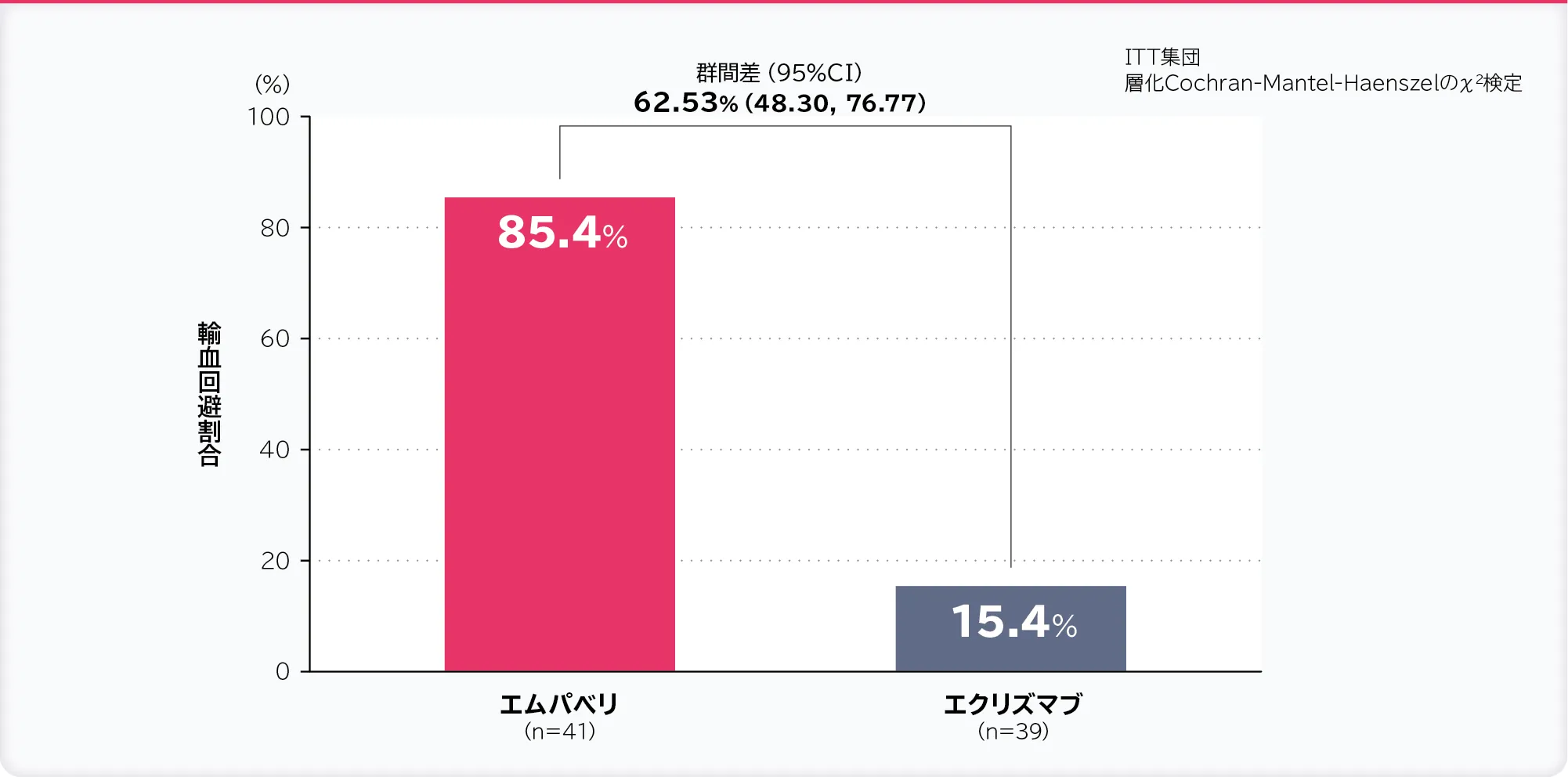
2. 輸血回避割合:無作為化投与期間内に輸血を必要としなかった被験者の割合〔重要な副次評価項目〕
輸血回避割合はエムパベリ群85.4%、エクリズマブ群15.4%、群間差は62.53%(95% CI:48.30, 76.77)であり、95%CIの下限値が非劣性マージンの−20%より大きかったため、エムパベリ群のエクリズマブ群に対する非劣性が示されました。
3. 無作為化投与期間の16週時点の網赤血球数のベースラインからの変化量〔重要な副次評価項目〕
無作為化投与期間の16週時点の網赤血球数のベースラインからの変化量の調整平均(最⼩⼆乗平均値)はエムパベリ群−135.82×109/L、エクリズマブ群27.79×109/L、群間差は−163.61×109/L(95%CI:−189.91, −137.30)であり、95%CIの上限値が非劣性マージンの10×109/Lより小さかったため、エムパベリ群のエクリズマブ群に対する非劣性が示されました。
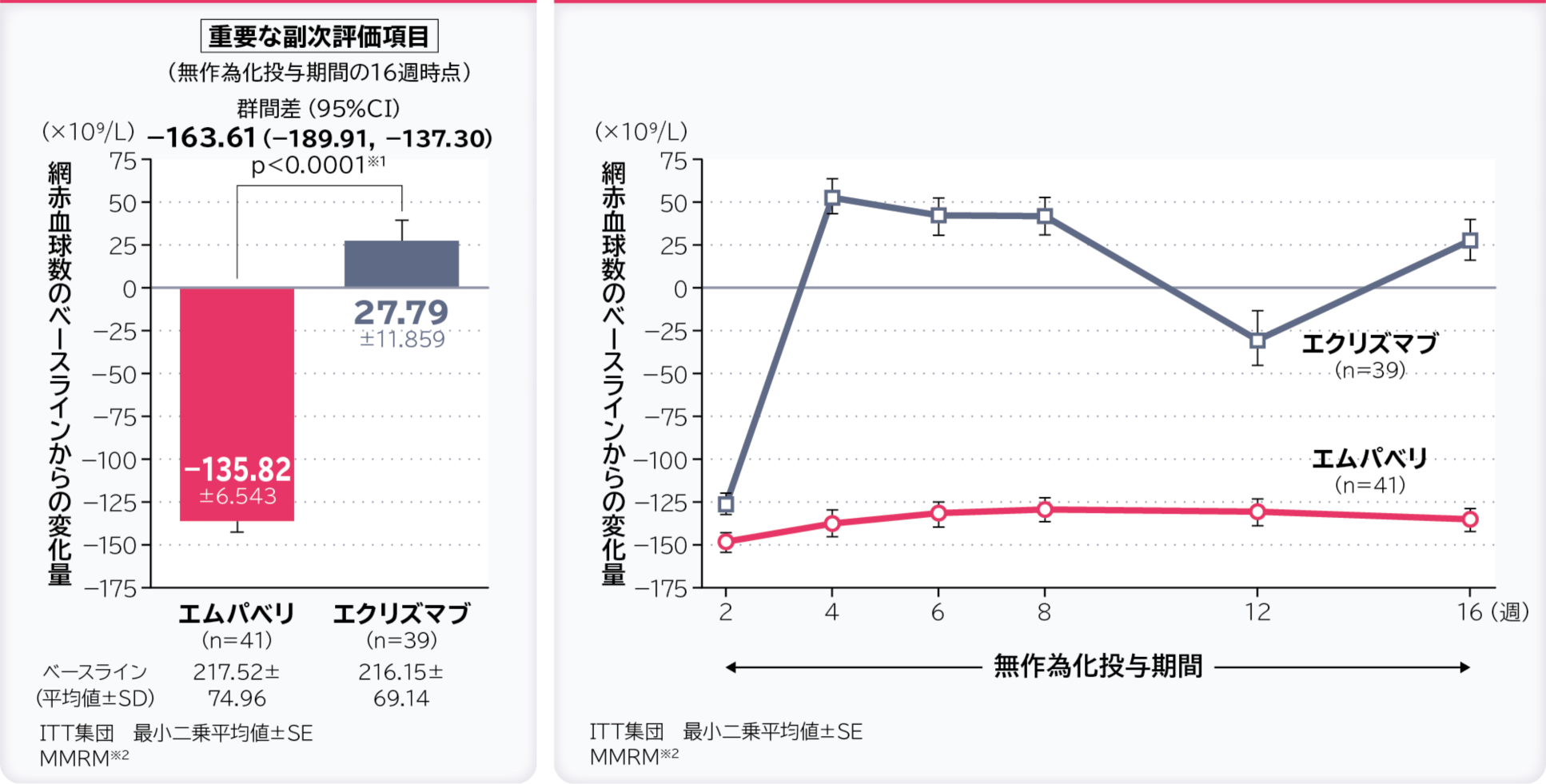
- ※1:有意水準α=0.05
- ※2:ベースライン、層別因子、投与、来院及び投与群と来院の交互作用で調整したデータを解析した。
4. 無作為化投与期間の16週時点のLDH値のベースラインからの変化量〔重要な副次評価項目〕
無作為化投与期間の16週時点のLDH値のベースラインからの変化量の調整平均(最⼩⼆乗平均値)はエムパベリ群−14.76U/L、エクリズマブ群−10.12U/L、群間差は−4.63U/L(95%CI:−181.30, 172.04)であり、95%CIの上限値が非劣性マージンの20U/Lより大きかったため、エムパベリ群のエクリズマブ群に対する非劣性は示されませんでした。
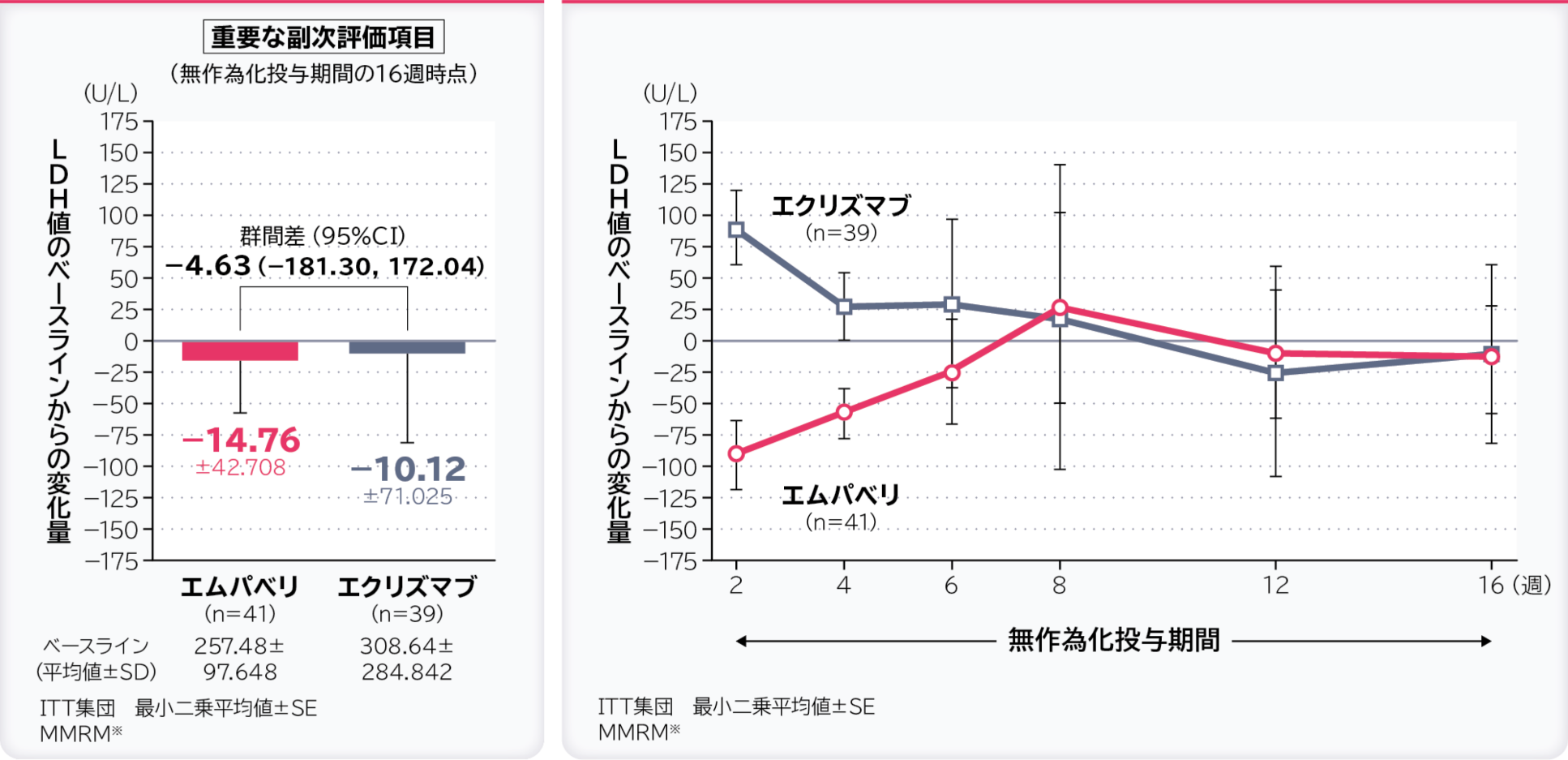
- ※:ベースライン、層別因子、投与、来院及び投与群と来院の交互作用で調整したデータを解析した。
5. 無作為化投与期間の16週時点のFACIT-Fatigueスコアのベースラインからの変化量〔重要な副次評価項目〕
注)
参考情報
- 注)事前に規定していた階層的検定手順に従い、非劣性の検証は実施しなかった。
- 注)事前に規定していた階層的検定手順に従い、非劣性の検証は実施しなかった。
無作為化投与期間の16週時点のFACIT-Fatigueスコアのベースラインからの変化量の調整平均(最⼩⼆乗平均値)はエムパベリ群9.22、エクリズマブ群-2.65、群間差は11.87(95%CI:5.49, 18.25)でした。
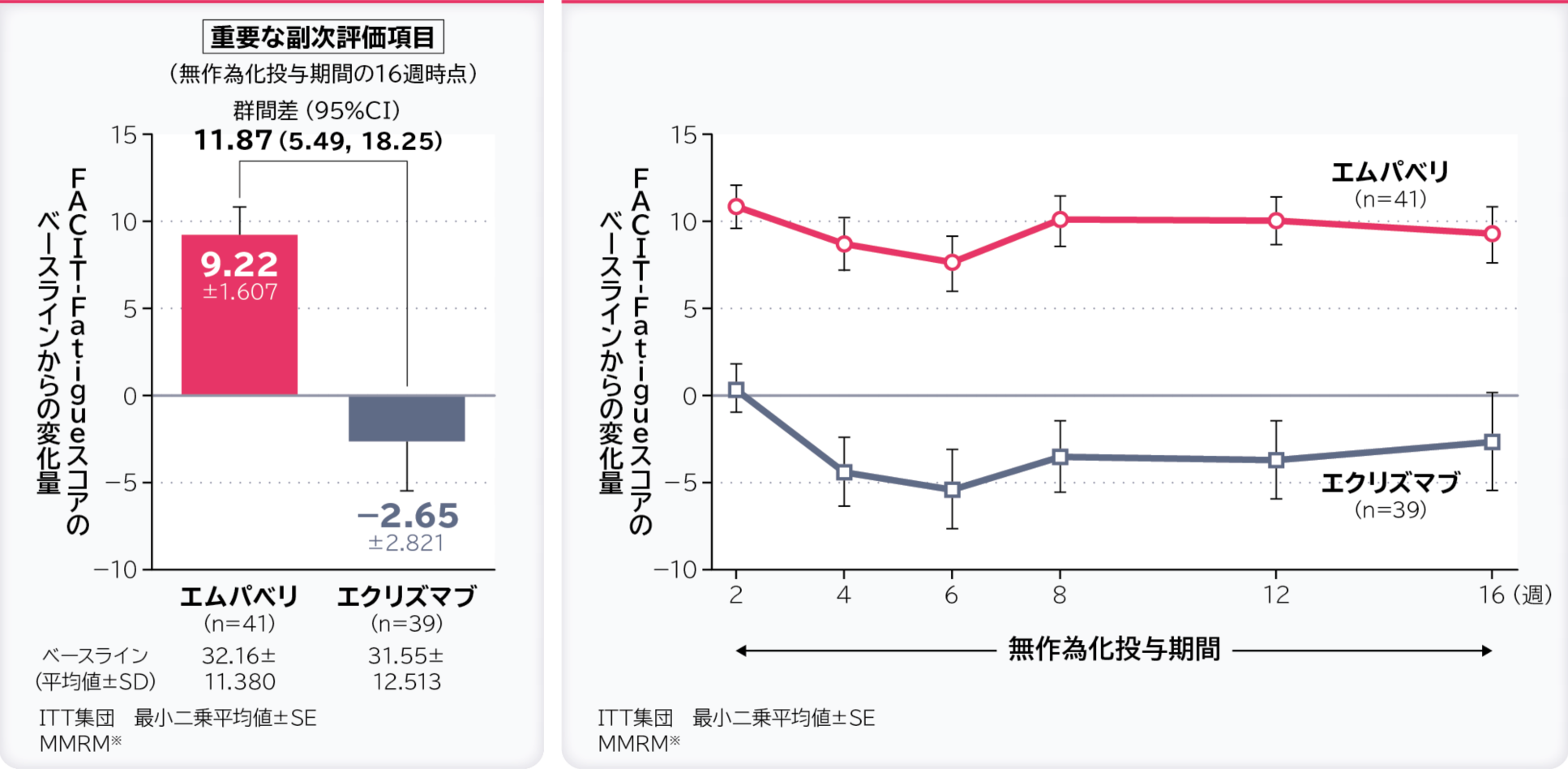
- ※:ベースライン、層別因子、投与、来院及び投与群と来院の交互作用で調整したデータを解析した。
6. 無作為化投与期間の16週時点の間接ビリルビン値のベースラインからの変化量〔その他の副次評価項目〕
無作為化投与期間の16週時点の間接ビリルビン値のベースラインからの変化量の調整平均(最⼩⼆乗平均値)はエムパベリ群−17.78μmol/L、エクリズマブ群4.15μmol/L、群間差は−21.93μmol/L(95%CI:−32.49, −11.36)でした。
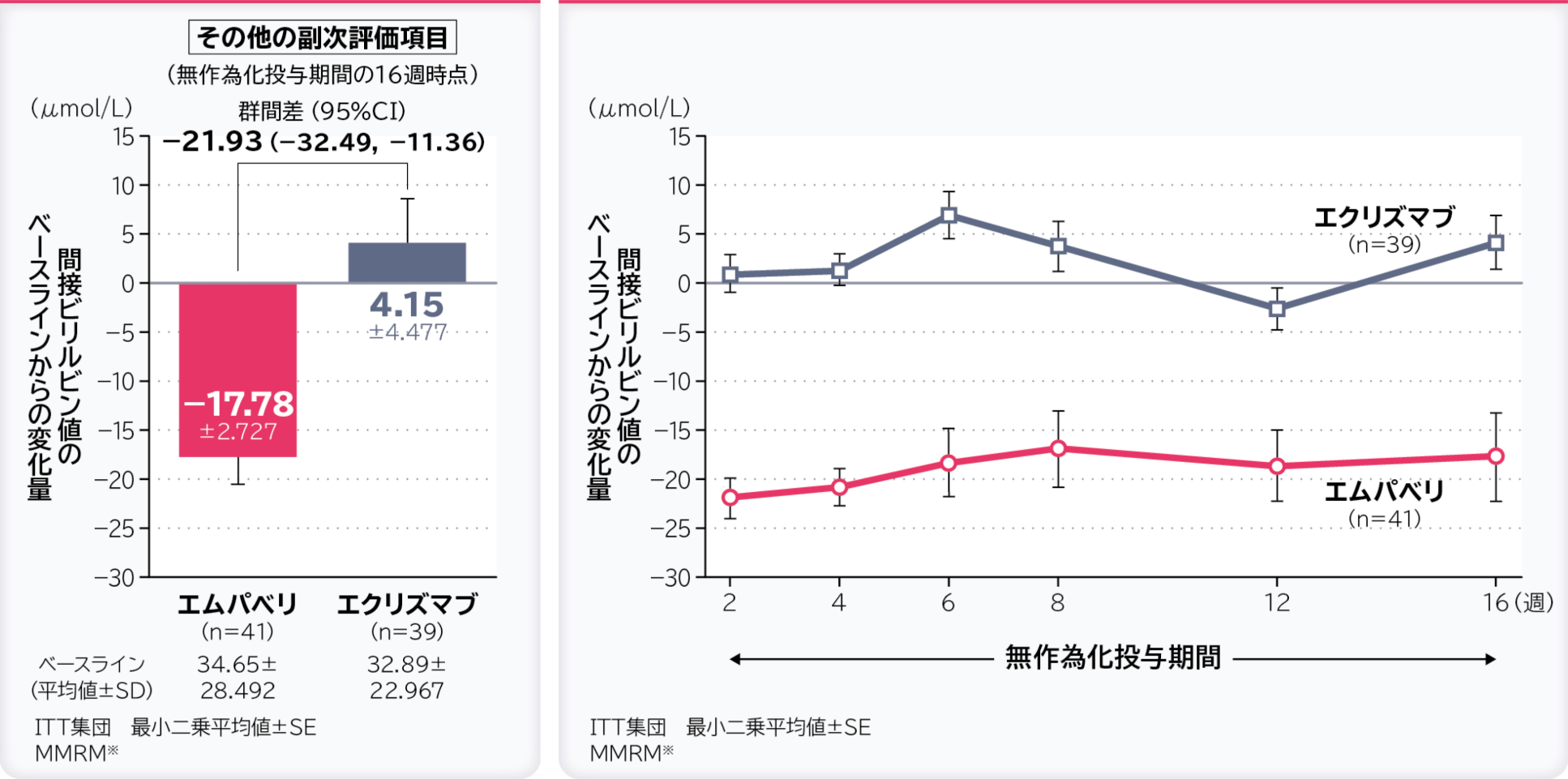
- ※:ベースライン、層別因子、投与、来院及び投与群と来院の交互作用で調整したデータを解析した。
安全性
[導入投与期間(4週間)] 全集団
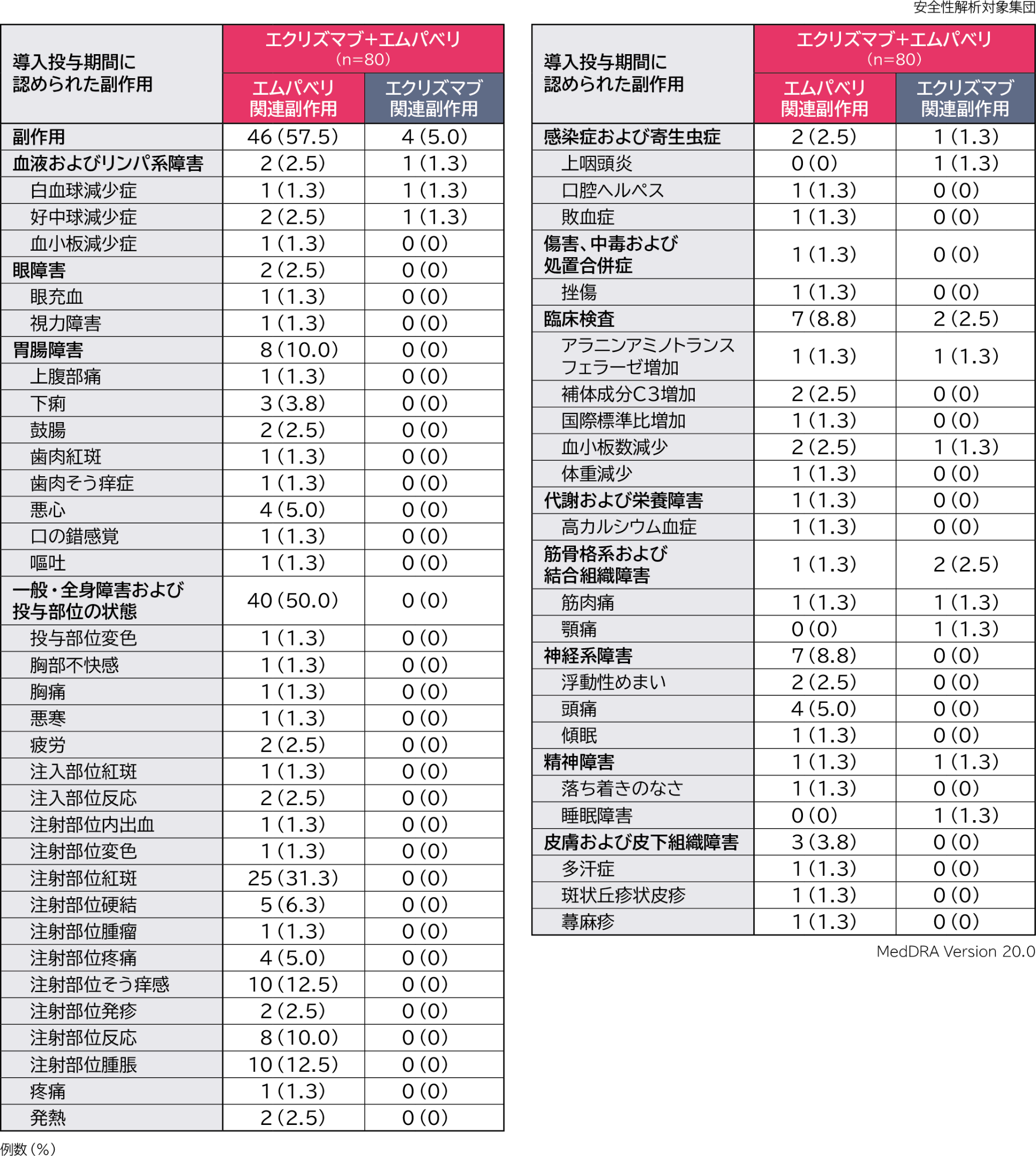
導入投与期間のエクリズマブ+エムパベリ併用投与における副作用は、エムパベリ関連副作用が80例中46例(57.5%)、エクリズマブ関連副作用が80例中4例(5.0%)に認められました。主なエムパベリ関連副作用(発現率5%以上)は注射部位紅斑25例(31.3%)、注射部位そう痒感、注射部位腫脹各10例(12.5%)、注射部位反応8例(10.0%)、注射部位硬結5例(6.3%)、悪心、注射部位疼痛、頭痛各4例(5.0%)、エクリズマブ関連副作用は白血球減少症、好中球減少症、上咽頭炎、アラニンアミノトランスフェラーゼ増加、血小板数減少、筋肉痛、顎痛、睡眠障害各1例(1.3%)でした。
重篤な副作用はエムパベリ関連で1例(敗血症)認められました。
投与中止に至った副作用は認められませんでした。
[無作為化投与期間(16週間)] 全集団
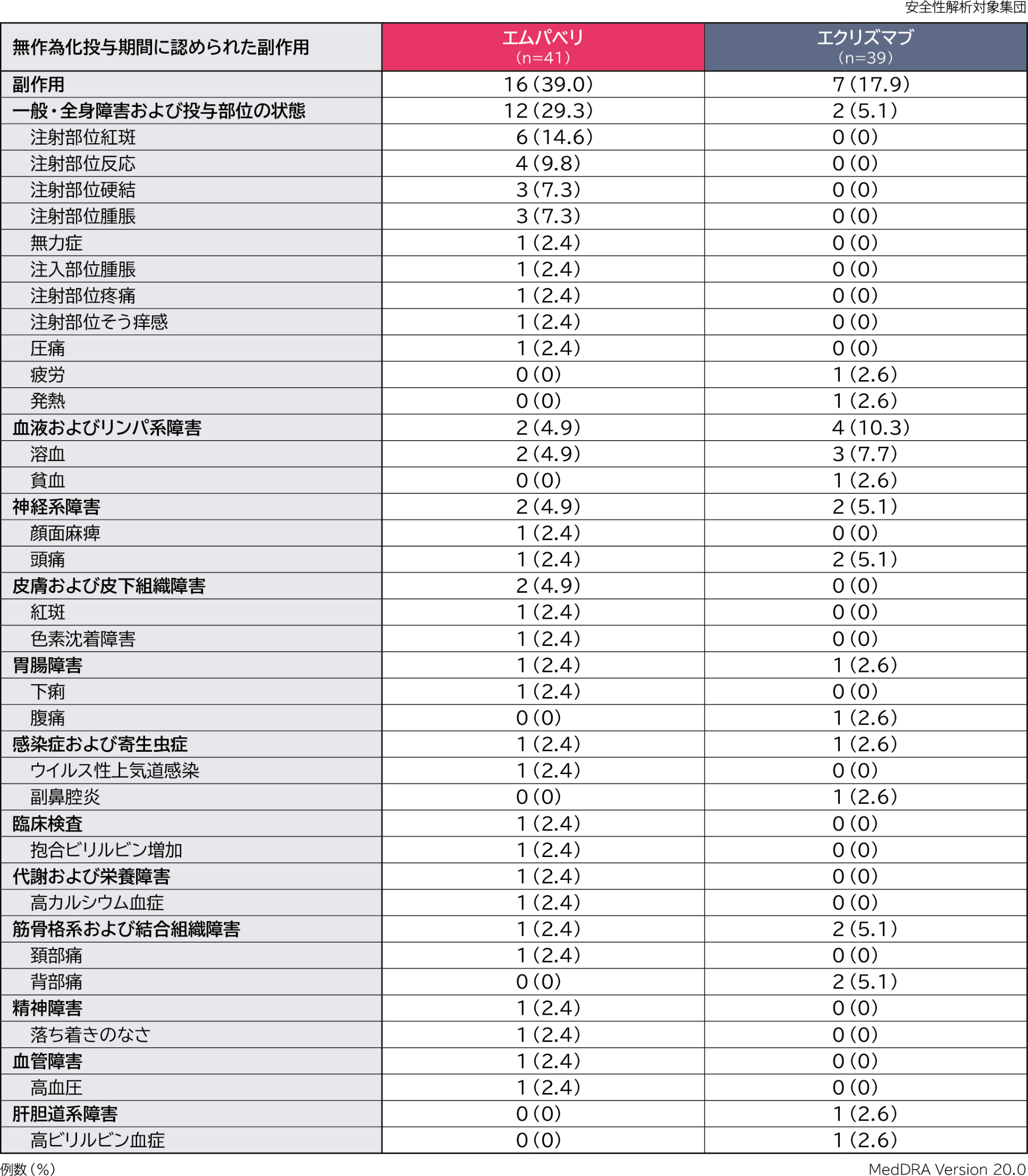
無作為化投与期間の副作用はエムパベリ群41例中16例(39.0%)、エクリズマブ群39例中7例(17.9%)に認められました。主な副作用(いずれかの群で発現率5%以上)はエムパベリ群で注射部位紅斑6例(14.6%)、注射部位反応4例(9.8%)、注射部位硬結、注射部位腫脹各3例(7.3%)、エクリズマブ群で溶血3例(7.7%)、頭痛、背部痛各2例(5.1%)でした。
重篤な副作用はエムパベリ群で1例(顔面麻痺)認められました。
投与中止に至った副作用はエムパベリ群で1例(溶血)認められました。
社内資料:外国⼈及び⽇本⼈PNH患者を対象とした国際共同第Ⅲ相試験(APL2-302試験)(2023年3⽉27⽇承認、申請資料概要2.7.6.9)(承認時評価資料)
Hillmen P, et al.:N Engl J Med 384:1028, 2021(PMID:33730455)
(本試験はApellis社の⽀援により⾏われた。)
製品に関するご不明な点は、
くすり相談窓口までお問い合わせください。
くすり相談窓口
受付時間:9:00〜17:45
(土日祝、休業日を除く)
当社は、日本製薬工業協会が提唱する
くすり相談窓口の役割・使命
に則り、
くすりの適正使用情報をご提供しています。