






























有効性
「警告、禁忌を含む使用上の注意」等は添付文書をご参照ください。
本剤は、国内第Ⅱ/Ⅲ相ブリッジング試験及び海外第Ⅲ相ブリッジング対象試験に基づき、海外臨床試験データを日本人に外挿することが可能であると判断され、臨床データパッケージに基づいて承認されました。このため、一部承認用法・用量と異なる成績が含まれますが、承認時評価資料のため掲載します。
国内第Ⅲ相臨床試験[HARUKA(LTS13618)試験]
関節リウマチ患者を対象としたDMARDs併用療法又は単剤療法試験
試験概要
■目的
■試験デザイン
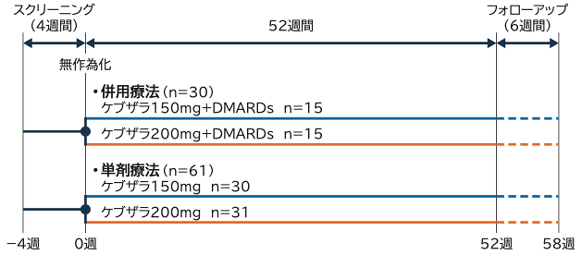
多施設共同、無作為化、二重盲検比較試験(国内第Ⅲ相臨床試験、52週間)
■対象
中等度から重度の活動性RA患者 91例
- 併用療法層:
- MTX以外のDMARDs(単剤又は併用療法)で効果不十分な患者 30例
- 単剤療法層:
- MTXに不耐、不適切又は効果不十分な患者 61例
■投与方法
- 併用療法層:
- 以下に示す2つの投与群に無作為に割り付け、ケブザラ150mg又は200mgを2週間に1回、52週間皮下投与した。また、各群とも基礎治療としてMTX以外のDMARDs(複数可)を投与した。
- 単剤療法層:
- 以下に示す2つの投与群に無作為に割り付け、ケブザラ150mg又は200mgを2週間に1回、52週間皮下投与した。
■評価項目
- 主要評価項目:
-
重篤な有害事象、特に注目すべき有害事象を含む有害事象、
臨床検査値(52週時)等
- 副次評価項目:
-
ACR2O改善率の経時変化
ベースラインからのDAS28-CRPの経時変化、DAS28-CRP寛解率
ベースラインからのHAQ-DIの経時変化等
■解析計画
安全性解析対象集団は、ランダム化された集団のうち、治験薬を少なくとも1回又は1回投与量の一部を投与されたすべての患者とした。有効性解析対象集団はmodified intent-to-treat(mITT)集団とし、observed case(OC)に基づき評価した。
有効性評価項目について検証的な解析は実施しなかった。
有効性評価項目の2値変数については、反応例数及び反応率を各療法層(併用療法層又は単剤療法層)の投与群別に示した。各評価時点での連続変数については、規定来院での評価時点における例数、平均値、標準偏差等を各療法層(併用療法層又は単剤療法層)の投与群別に示した。
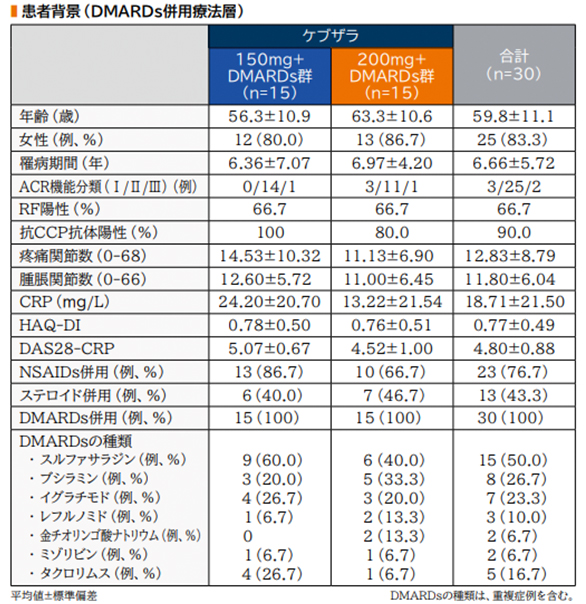
患者背景(DMARDs併用療法層)
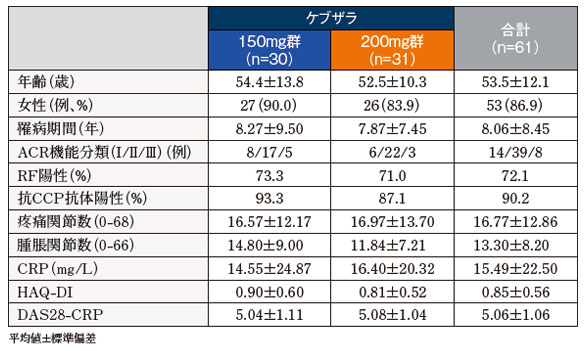
患者背景(ケブザラ単剤療法層)
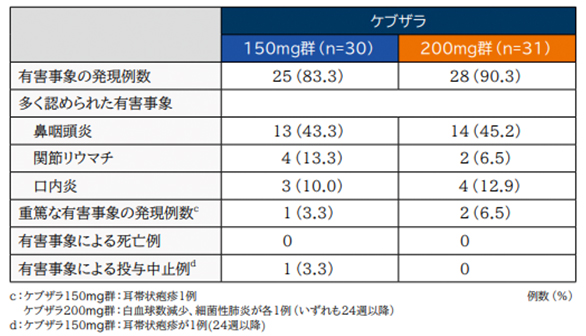
(1)安全性(52週時)[主要評価項目]
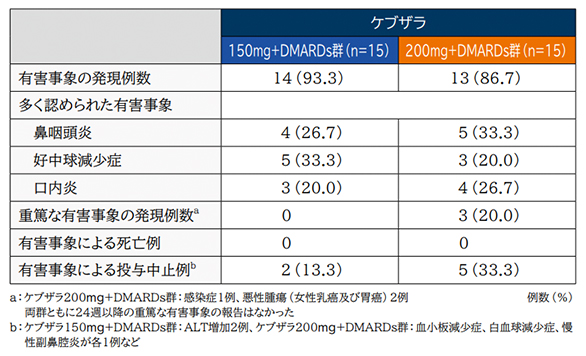
DMARDs併用療法層:
有害事象の発現率は、ケブザラ150mg+DMARDs群で93.3%(14/15例)、200mg+DMARDs群で86.7%(13/15例)であった。
主な有害事象は、鼻咽頭炎がそれぞれ26.7%(4/15例)、33.3%(5/15例)、好中球減少症33.3%(5/15例)、20.0%(3/15例)、口内炎20.0%(3/15例)、26.7%(4/15例)であった。特に注目すべき有害事象として多く認められたものは、感染症及び白血球減少症であった。
重篤な有害事象は、24週時までにケブザラ200mg+DMARDs群で20.0%(3/15例)、3例4件(感染症1例、悪性腫瘍2例)に認められ、150mg+DMARDs群では認められず、24週以降の重篤な有害事象の報告はなかった。また、ケブザラ200mg+DMARDs群の悪性腫瘍(女性乳癌及び胃癌)2例において、本剤との因果関係は否定された。
投与中止に至った有害事象は、ケブザラ150mg+DMARDs群で13.3%(2/15例)にALT増加2例、200mg+DMARDs群で33.3%(5/15例)に血小板減少症、白血球減少症、慢性副鼻腔炎が各1例認められた。なお、死亡例は報告されなかった。
ケブザラ単剤療法層:
有害事象の発現率は、ケブザラ150mg群で83.3%(25/30例)、200mg群で90.3%(28/31例)であった。
多く認められた有害事象は鼻咽頭炎がそれぞれ43.3%(13/30例)、45.2%(14/31例)、関節リウマチ13.3%(4/30例)、6.5%(2/31例)、口内炎10.0%(3/30例)、12.9%(4/31例)であった。特に注目すべき有害事象として多く認められたものは、両群ともに感染症であった。
重篤な有害事象はケブザラ150mg群で耳帯状疱疹1例、200mg群で白血球数減少、細菌性肺炎が各1例に認められ、投与中止に至った有害事象はケブザラ150mg群で耳帯状疱疹1例が認められたが、これらはすべて24週以降に認められた。死亡例は報告されなかった。
(2)ACR20改善率の推移[副次評価項目]
ACR20改善率は、以下の通りであった。
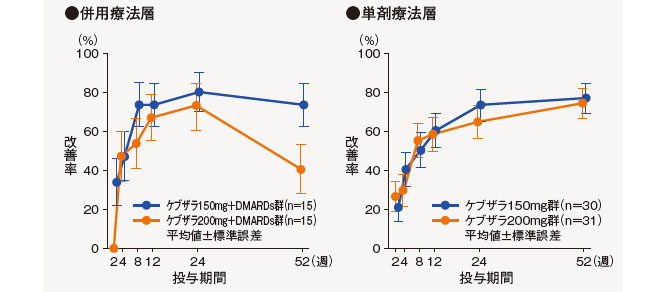
中止(救済治療を含む)など判定不能な患者について、それ以降の時点には効果不十分症例として補完した。
(3)DAS28-CRP変化量の推移[副次評価項目]
ベースラインからのDAS28-CRP変化量は、以下の通りであった。
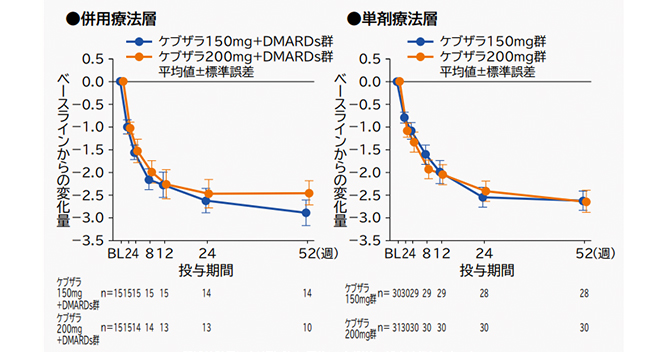
記述統計量により群ごとに要約し、欠測値の補完は行わなかった。
(4)DAS28-CRP寛解率の推移[副次評価項目]
DAS28-CRP寛解の基準(2.6未満)に達した患者の割合は、以下の通りであった。
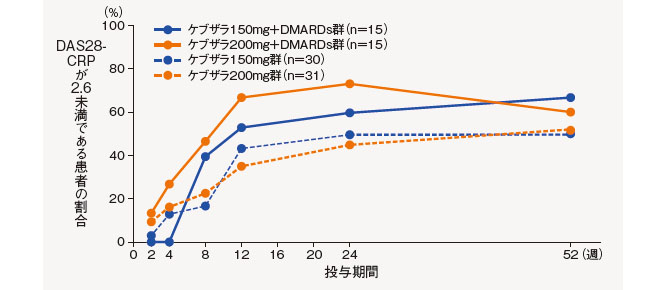
中止(救済治療を含む)など判定不能な患者について、それ以降の時点には効果不十分症例として補完した。
(5)HAQ-DI変化量の推移[副次評価項目]
ベースラインからのHAQ-DI変化量は、以下の通りであった。
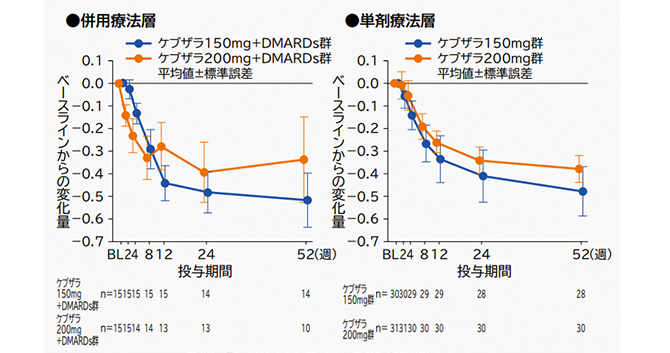
記述統計量により群ごとに要約し、欠測値の補完は行わなかった。
社内資料:国内第Ⅲ相二重盲検比較試験(メトトレキサート以外のDMARDs併用又は単剤投与試験)[LTS13618試驗](承認時評価資料)
製品に関するご不明な点は、
くすり相談窓口までお問い合わせください。
くすり相談窓口
受付時間:9:00〜17:45
(土日祝、休業日を除く)
当社は、日本製薬工業協会が提唱する
くすり相談窓口の役割・使命
に則り、
くすりの適正使用情報をご提供しています。