






























有効性
「警告、禁忌を含む使用上の注意」等は添付文書をご参照ください。
本剤は、国内第Ⅱ/Ⅲ相ブリッジング試験及び海外第Ⅲ相ブリッジング対象試験に基づき、海外臨床試験データを日本人に外挿することが可能であると判断され、臨床データパッケージに基づいて承認されました。このため、一部承認用法・用量と異なる成績が含まれますが、承認時評価資料のため掲載します。
海外第Ⅲ相臨床試験[TARGET(EFC10832)試験](海外データ)
TNF阻害薬で効果不十分な関節リウマチ患者を対象としたDMARDs併用試験
試験概要
■目的
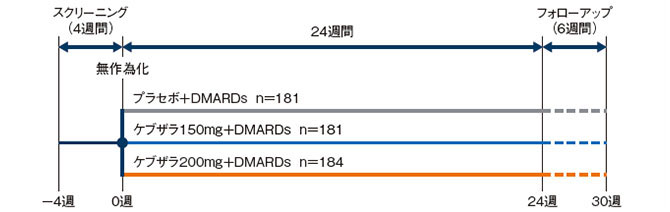
■試験デザイン
多施設共同、無作為化、プラセボ対照二重盲検、並行群間比較試験(海外第Ⅲ相臨床試験、24週間)
■対象
TNF阻害薬で効果不十分又は不耐の中等度から重度の活動性RA患者 546例
■投与方法
以下に示す3つの投与群に無作為に割り付け、いずれも生物学的製剤以外のDMARDs併用下で2週間に1回、24週間皮下投与した。
・プラセボ2週間に1回 181例
・ケブザラ150mg 2週間に1回 181例
・ケブザラ200mg 2週間に1回 184例
- 救済治療:
-
12週以降、効果不十分の定義に該当する患者は、非盲検の継続投与試験に移行し救済治療を可能とした。
[腫脹関節数又は圧痛関節数の改善が2回連続して(少なくとも4週間あけて)ベースラインから20%未満]
■評価項目
- 主要評価項目:
-
ACR20改善率(24週時)
HAQ-DI変化量(12週時)(いずれも検証的な解析項目)
- 副次評価項目:
- ACR20/50/70改善率(12週時)、ACRコアセットの変化量(12、24週時)、ACR20改善率の推移 等
- 安全性:
- 重篤な有害事象、注目すべき有害事象を含む有害事象、臨床検査値 等
■解析計画
試験の目標症例数はHAQ-DIの評価項目においてプラセボ群に対する差を検出できるよう設計した。
主要な有効性解析対象集団はintent-to-treat(ITT)集団とした。
主要評価項目のうちACR20改善率(24週時)は、地域及び前治療としてのTNF阻害薬の薬剤数を層とした両側Cochran-Mantel-Haenszel検定による解析を行い、ケブザラの各用量群とプラセボ群との間で対比較を行った。ベースラインからのHAQ-DI変化量(12週時)は、ベースラインを共変量とし、投与群、地域、前治療としてのTNF阻害薬の薬剤数、時点及び投与群と時点の交互作用を因子としたmixed model repeated measureS(MMRM)を用いて解析した。
全体の第1種の過誤確率を5%に制御するため、2つの主要評価項目について、各用量群において多重性の調整を適用した。多重性の調整は、評価項目の記載順序の通りに閉手順を用いて行った。
副次評価項目のうち2値変数については、ACR20改善率と同様に両側Cochran-Mantel-Haenszel検定を用いて解析し、連続変数についてはHAQ-DIと同じ手法で解析した。
患者背景
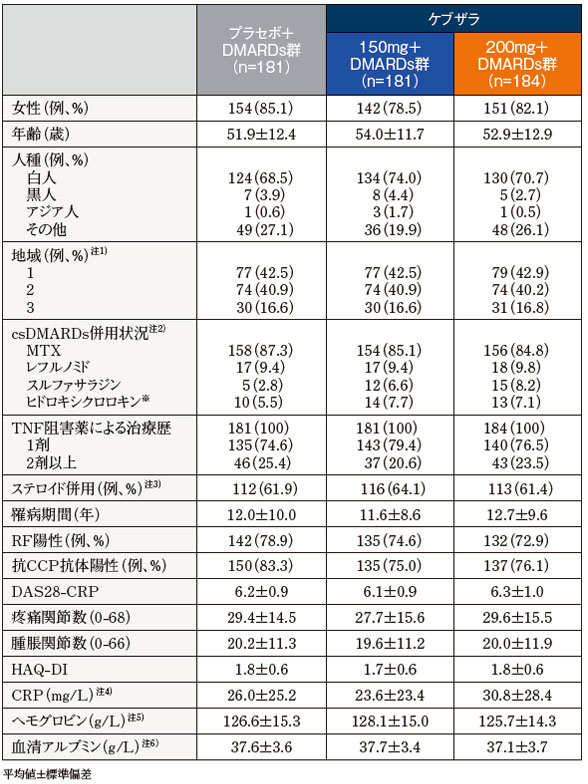
- 注1)
-
- 地域1:
- オーストラリア、カナダ、チェコ、ドイツ、ギリシャ、ハンガリー、イスラエル、イタリア、ニュージーランド、ポルトガル、スペイン、米国
- 地域2:
- アルゼンチン、ブラジル、チリ、コロンビア、エクアドル、グアテマラ、メキシコ、ペルー
- 地域3:
- リトアニア、ポーランド、ロシア、韓国、台湾、タイ、トルコ、ウクライナ
- 注2)
- 6.4%の患者が2種、0.7%の患者が3種のcsDMARSs(従来型合成抗リウマチ薬)を併用していたと報告された。
- 注3)
- 経ロステロイドの投与は、無作為化の4週以上前からプレドニゾロン換算で10mg/日以下であれば可能であったが、投与量は有害事象が発現した場合を除き変更が認められなかった。
- 注4)
- 試験を実施した地域では、50歳女性の正常範囲は3.1mg/L未満であった。
- 注5)
- 試験を実施した地域では、50歳男性の正常範囲は135-175g/L、50歳女性では120-160g/Lであった。
- 注6)
- 試験を実施した地域では、50歳女性の正常範囲は35-55g/Lであった。
※関節リウマチに対しては本邦未承認
(1)ACR20改善率(24週時)[主要評価項目]
24週時におけるACR20改善率は、ケブザラ2用量群のいずれにおいてもプラセボ+DMARDs群に対して優越性が検証され、ケブザラ150mg+DMARDs群で55.8%、ケブザラ200mg+DMARDs群で60.9%であった。
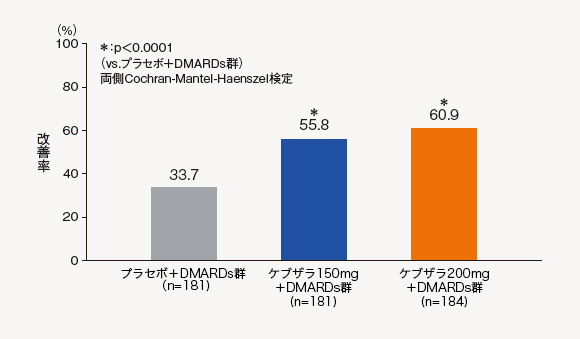
中止(救済治療を含む)など判定不能な患者について、それ以降の時点には効果不十分症例として補完した。
(2)ACR20改善率の推移(24週)[副次評価項目]
ACR20改善率の推移は、以下の通りであった。
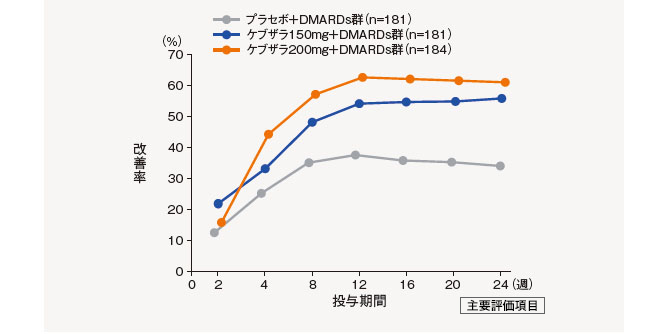
中止(救済治療を含む)など判定不能な患者について、それ以降の時点には効果不十分症例として補完した。
(3)HAQ-DI変化量(12週時)[主要評価項目]
12週時におけるベースラインからのHAQ-DI変化量は、ケブザラ2用量群のいずれにおいてもプラセボ+DMARDs群に対して優越性が検証された。
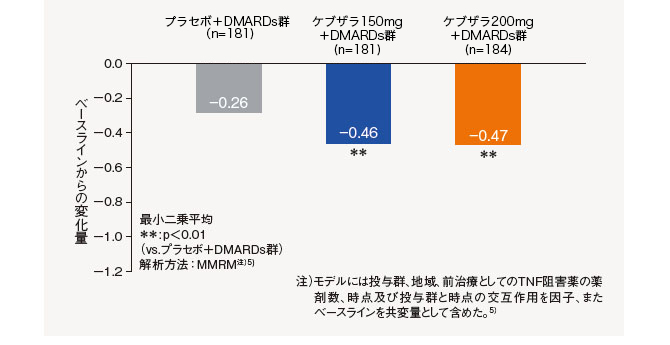
社内資料:海外第Ⅲ相プラセボ対照二重盲検比較試験(生物学的製剤以外のDMARDs併用試験)[EFC10832試験](承認時評価資料)
(4)安全性
有害事象の発現率は、ケブザラ150mg+DMARDs群で65.7%(119/181例)、200mg+DMARDs群で65.2%(120/184例)、プラセボ+DMARDs群で49.7%(90/181例)であった。
多く認められた有害事象は、好中球減少症がそれぞれ12.7%(23/181例)、12.5%(23/184例)、1.1%(2/181例)、次いで鼻咽頭炎が6.1%(11/181例)、3.8%(7/184例)、5.0%(9/181例)、尿路感染症が3.3%(6/181例)、7.1%(13/184例)、6.6%(12/181例)であった。
重篤な有害事象は、ケブザラ150mg+DMARDs群3.3%(6/181例)、200mg+DMARDs群5.4%(10/184例)、プラセボ+DMARDs群3.3%(6/181例)に認められた。最も多く認められたものは感染症であり、ケブザラ150mg+DMARDs群1例、200mg+DMARDs群2例、プラセボ十DMARDs群2例であった。また、非播種性の帯状疱疹が3例(ケブザラ200mg+DMARDs群2例、プラセボ+DMARDs群1例)に認められた。
投与中止に至った有害事象は、ケブザラ150mg+DMARDs群7.7%(14/181例)、200mg+DMARDs群9.2%(17/184例)、プラセボ+DMARDs群4.4%(8/181)に認められ、主なものは好中球減少症であった。死亡例はプラセボ+DMARDs群の1例に認められた。
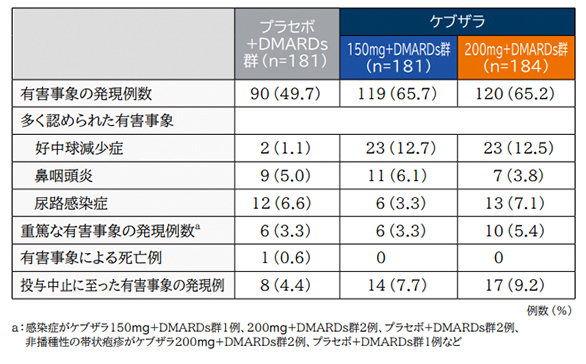
社内資料:海外第Ⅲ相プラセボ対照二重盲検比較試験(生物学的製剤以外のDMARDs併用試験)[EFC10832試驗](承認時評価資料)
Fleischmann R et al. Arthritis Rheumatol 2017;69(2):277-290
本試験はSanofi の出資により実施された。
Copyright 2017 Wiley. Used with permission from Fleischmann R Sarilumab and Nonbiologic Disease-Modifying Antirheumatic Drugs in Patients With Active Rheumatoid Arthritis and Inadequate Response or Intolerance to Tumor Necrosis Factor Inhibitors, Arthritis Rheumatol, Wiley Periodicals, Inc.
製品に関するご不明な点は、
くすり相談窓口までお問い合わせください。
くすり相談窓口
受付時間:9:00〜17:45
(土日祝、休業日を除く)
当社は、日本製薬工業協会が提唱する
くすり相談窓口の役割・使命
に則り、
くすりの適正使用情報をご提供しています。