





























- クレセンバ®トップ
- セミナー・講演会
- D-indexによる真菌感染症リスク計算ツール
- クレセンバ®について
- 資料ライブラリ
- 薬理動画
- クレセンバ座談会
- 血液内科領域における抗真菌感染症戦略
- どうする?NTM×CPA!
- ガイドライン
- JAID/JSC感染症治療ガイド2023
- 白血病および造血細胞移植患者における侵襲性糸状菌感染症の治療推奨
- 製品情報topics
- 慢性呼吸器疾患に潜む真菌感染
- エキスパートに聞くシリーズ
- クレセンバ製品詳細

- クレセンバ®トップ
- セミナー・講演会
- D-indexによる真菌感染症リスク計算ツール
- クレセンバ®について
- 資料ライブラリ
- 薬理動画
- クレセンバ座談会
- 血液内科領域における抗真菌感染症戦略
- どうする?NTM×CPA!
- ガイドライン
- JAID/JSC感染症治療ガイド2023
- 白血病および造血細胞移植患者における侵襲性糸状菌感染症の治療推奨
- 製品情報topics
- 慢性呼吸器疾患に潜む真菌感染
- エキスパートに聞くシリーズ
国内第III相試験
日本人深在性真菌症患者を対象とした臨床試験
(AK1820-301試験)1), 2)
結果
(1)投与開始から投与終了後35日目までに有害事象を発現した患者の割合(安全性解析対象集団)【主要評価項目】
●コホートA
慢性肺アスペルギルス症における有害事象の発現頻度は、クレセンバ群90.4%(47/52例) 、ボリコナゾール群92.6%(25/27例)であった。
侵襲性アスペルギルス症における有害事象の発現頻度は、クレセンバ群3/3例、ボリコナゾール群1/1例であった。
●コホートB
ムーコル症における有害事象の発現頻度は3/3例、クリプトコックス症における有害事象の発現頻度は90.0%(9/10例)であった。
(2)42日目、84日目及びEOTの総合効果総合効果(DRC判定)(mITT集団)【副次評価項目】
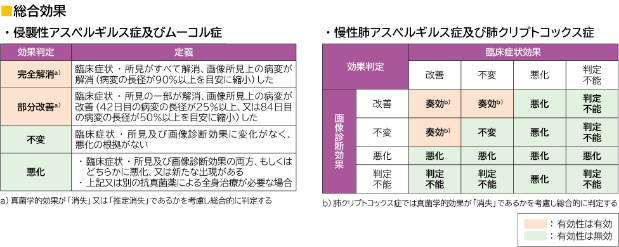
有効性の判定基準については下部に記載
●コホートA
1)慢性肺アスペルギルス症
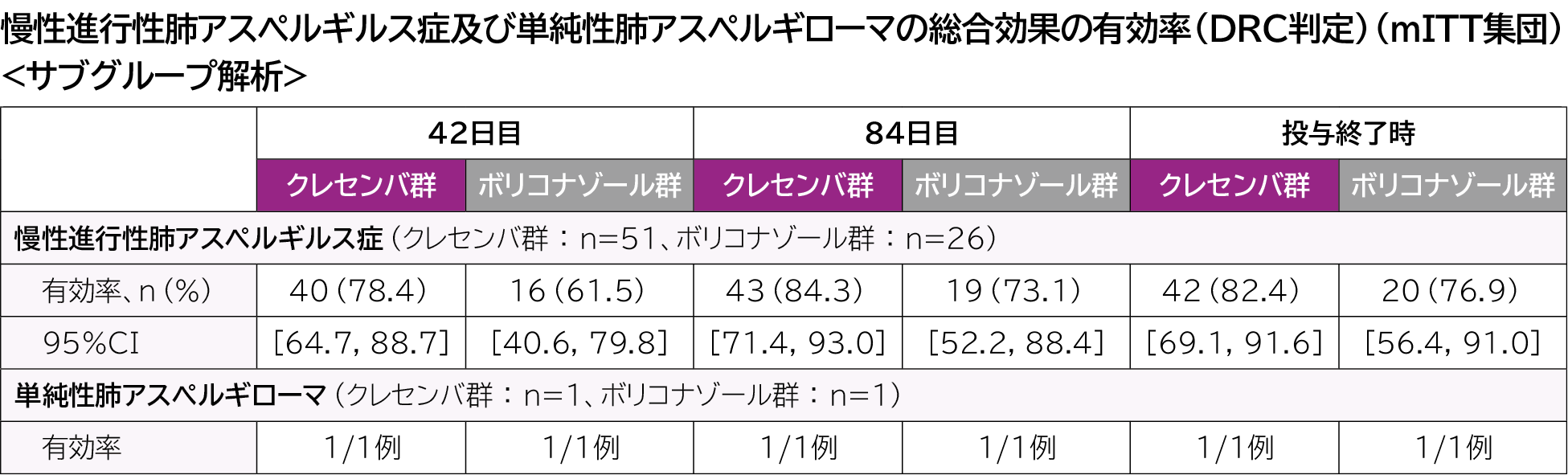
■ 42日目、84日目及び投与終了時の総合効果(DRC判定)
総合効果の有効率は、クレセンバ群及びボリコナゾール群でそれぞれ、42日目では78.8%及び63.0%、84日目では84.6%及び74.1%、投与終了時では82.7%及び77.8%であった。

このうち、慢性進⾏性肺アスペルギルス症及び単純性肺アスペルギローマの総合効果は以下のとおりであった。
2)侵襲性アスペルギルス症
■ 42日目、84日目及び投与終了時の総合効果(DRC判定)
総合効果は、42日目及び投与終了時でクレセンバ群1/3例、ボリコナゾール群1/1例が有効と判定された。クレセンバ群及びボリコナゾール群で有効と判定された患者は、それぞれ62日目、37日目に投与中止した。

●コホートB
1)ムーコル症
■ 42日目、84日目及び投与終了時の総合効果(DRC判定)
総合効果は、42日目、84日目及び投与終了時のいずれの時点においても、クレセンバ群1/3例が有効と判定された。

2)クリプトコックス症
クリプトコックス症10例は、全例が肺クリプトコックス症であった。
■ 42日目、84日目及び投与終了時の総合効果(DRC判定)
総合効果の有効率は、42日目、84日目及び投与終了時でいずれの時点においても90.0%であった。

安全性(安全性解析対象集団)
●コホートA
副作用は、クレセンバ群60.0%(36/60例)、ボリコナゾール群80.0%(24/30例)に認められた。主な副作用は、クレセンバ群では肝機能検査値上昇5例(8.3%)、肝機能異常、ほてり各4例(6.7%)、悪心3例(5.0%)であり、ボリコナゾール群では肝機能異常、羞明各7例(23.3%)、視力障害4例(13.3%)、肝機能検査値上昇、色覚異常各3例(10.0%)、肝障害、悪⼼、嘔吐、浮動性めまい、霧視、幻視各2例(6.7%)であった。
重篤な副作用は、クレセンバ群4例、ボリコナゾール群1例に認められ、その内訳はクレセンバ群では胆嚢炎、筋力低下、抗利尿ホルモン不適合分泌、低ナトリウム血症各1例、ボリコナゾール群では薬物性肝障害、横紋筋融解症各1例であった。
投与中止に至った副作用は、クレセンバ群8例、ボリコナゾール群4例に認められ、その内訳は、クレセンバ群では肝機能異常2例、高カリウム血症、低ナトリウム血症、感覚鈍麻、肝機能検査値上昇、悪心、注射部位蕁麻疹、腎機能障害、呼吸困難、口腔咽頭不快感、冷汗各1例であり、ボリコナゾール群では肝機能異常、薬物性肝障害、多発ニューロパチー、視力障害、横紋筋融解症各1例であった。
本試験のコホートAにおいて、死亡に至った副作用は両群で認められなかった。
●コホートA/B併合(クレセンバ群)
副作用は60.3%(44/73例)に認められ、主な副作用は、肝機能検査値上昇6例(8.2%)、肝機能異常、悪⼼各5例(6.8%)、ほてり4例(5.5%)であった。
重篤な副作⽤は5例に認められ、コホートAの事象に加え、死亡1例が認められた。
投与中⽌に⾄った副作⽤は9例に認められ、コホートAの事象に加え、⼼電図異常1例が認められた。
本試験のコホートA/B併合において、死亡に至った副作用が1例(コホートB)に認められ、発現事象は「死亡」であった。当該事象は突然死であり、剖検未実施で合理的に説明し得る死因の特定が困難であったため、治験薬との因果関係は否定されなかった。

試験概要
| 目的 | 日本人深在性真菌症患者を対象に、クレセンバを静脈内投与又は経口投与した場合の安全性及び有効性を検討する。 |
|---|---|
| 試験デザイン |
コホートA:多施設共同、無作為化、非盲検、実薬対照試験
コホートB:多施設共同、非盲検、非対照試験 |
| 対象 |
コホートA:慢性肺アスペルギルス症(慢性進行性肺アスペルギルス症、単純性肺アスペルギローマ)及び侵襲性アスペルギルス症患者90例(慢性肺アスペルギルス症:クレセンバ群52例、ボリコナゾール群27例、侵襲性アスペルギルス症:クレセンバ群3例、ボリコナゾール群1例、No deep mycosis(DRCが深在性真菌症の診断基準に合致しない、または可能性例と判定した症例):クレセンバ群5例、ボリコナゾール群2例)
コホートB:ムーコル症及びクリプトコックス症(肺クリプトコックス症、播種性クリプトコックス症、クリプトコックス脳髄膜炎)患者13例(ムーコル症3例、クリプトコックス症10例;いずれもクレセンバ群)
【主な選択基準】
下記と診断された20歳以上の患者
【主な除外基準】
#:国内未承認 |
| 投与方法 |
治験薬を最大84日間投与した。
治験薬の投与方法

|
| 評価項目 |
[主要評価項目]
[副次評価項目]
[安全性評価項目]
|
| 解析計画 |
解析対象集団
mITT集団を主たる有効性解析対象集団とした。
解析方法
[主要評価項目]
※ DRC判定による総合効果の各評価時点で欠測が生じた場合は無効、全死因死亡率の各評価時点での生存状況が不明であった場合は死亡と扱った。 |
- DRC:治験依頼者及び治験責任医師とは独立した感染症専門医及び放射線専門医から構成されるデータレビュー委員会。
効果判定基準
1)承認時評価資料, 社内資料:日本人深在性真菌症を対象とした国内第Ⅲ相試験(AK1820-301試験)
2) Kohno S et al. J Infect Chemother. 2022; S1341-321X(22)00293-8. doi: 10.1016/j.jiac.2022.10.010.
[Epub ahead of print] 本試験は旭化成セラピューティクス株式会社の支援により実施された。
- 4. 効能・効果
- 下記の真菌症の治療
- 〇アスペルギルス症(侵襲性アスペルギルス症、慢性進行性肺アスペルギルス症、単純性肺アスペルギローマ)、〇ムーコル症、〇クリプトコックス症(肺クリプトコックス症、播種性クリプトコックス症(クリプトコックス脳髄膜炎を含む))
- 7. 用法・用量に関連する注意
- 7.1 カプセル剤と注射剤は医師の判断で切り替えて使用することができる。
- 7.2 投与期間は基礎疾患の状態、免疫抑制からの回復及び臨床効果に基づき設定すること。
製品に関するご不明な点は、
くすり相談窓口までお問い合わせください。
くすり相談窓口
受付時間:9:00〜17:45
(土日祝、休業日を除く)
当社は、日本製薬工業協会が提唱する
くすり相談窓口の役割・使命
に則り、
くすりの適正使用情報をご提供しています。
本剤は、海外臨床試験データを日本人に外挿することが可能であると判断され、国内第Ⅲ相試験及び海外第Ⅲ相試験を含む臨床データパッケージに基づいて承認されました。このため、一部承認外の菌種による真菌症の成績が含まれますが、承認時評価資料のため掲載します。