





























- プラケニル®トップ
- セミナー・講演会
- プラケニル®について
- SLEについて
- 評価指標(表計算)
- 実践的SLE診療の How to ~ 患者コミュニケーションから紐解く
- 症例からみるSLEの初診から確定診断までの経過
- エキスパートからのメッセージ
- Lupus Research Outlook Today
- 資料ライブラリ
- Web講演会記録集
- Lupus Expert’s View 2021 ~長期予後を考えたSLE診療~
- プラケニル座談会
- EULAR recommendations 2023
- SLE・プラケニル患者説明動画
- プラケニル製品詳細

- プラケニル®トップ
- セミナー・講演会
- プラケニル®について
- SLEについて
- 評価指標(表計算)
- 実践的SLE診療の How to ~ 患者コミュニケーションから紐解く
- 症例からみるSLEの初診から確定診断までの経過
- エキスパートからのメッセージ
- Lupus Research Outlook Today
- 資料ライブラリ
- Web講演会記録集
- Lupus Expert’s View 2021 ~長期予後を考えたSLE診療~
- プラケニル座談会
- EULAR recommendations 2023
- SLE・プラケニル患者説明動画
第Ⅲ相試験(有効性)
活動性皮膚病変を有するCLE患者(SLEの合併の有無を問わない)を対象とした国内第Ⅲ相試験(EFC12368試験)6)
試験概要
目的:
ステロイド剤併用/非併用下で活動性皮膚病変を有する(CLASI活動性スコア4点以上)皮膚エリテマトーデス(CLE)および全身性エリテマトーデス(SLE)患者において、プラケニル(ヒドロキシクロロキン硫酸塩)1日1回投与時の16週間での皮膚病変に対する有効性について検討する。
対象:
CLEと診断された日本人患者[SLEの合併の有無を問わない。ステロイド剤併用/非併用下で活動性皮膚病変を有する(CLASI活動性スコア4点以上)]
〔有効性解析対象集団(FAS)96例、安全性解析対象集団103例〕
最大の解析対象集団(Full Analysis Set:FAS)96例
[プラケニル/プラケニル群72例、プラセボ/プラケニル群24例]
無作為化された患者のうち、治験薬投与後に利用可能なCLASI 活動性スコアが存在しないなどの理由による重要な逸脱のない投与例とした。仮に割付けと異なった治験薬が投与されたとしても、割り付けられた投与群とみなすこととした。
安全性解析対象集団103例
[プラケニル/プラケニル群77例、プラセボ/プラケニル群26例]
無作為化され、治験薬を1回でも投与された(不完全な投与を含む)患者とした。
方法:
二重盲検、ベースライン対照、多施設共同試験
プラケニル200mgまたは400mg(6.5mg/kgを超えない)またはプラセボを二重盲検法により1日1回、16週間経口投与し、主要な有効性評価を行った。プラセボ群はプラケニル群との統計学的な比較を目的としない参照群として設定した。
その後、二重盲検期を終了したプラケニル群およびプラセボ群の全患者に対してプラケニル200mgまたは400mg(6.5mg/kgを超えない)を単盲検下で36週間継続投与し(投与後16週以降52週まで)、長期有効性および安全性を検討した。また、治験薬最終投与後に3週間の後観察期(投与後52週以降55週まで)を設けた。

評価項目:
〈有効性〉
主要評価項目 ベースライン時と治験薬投与後16週時点でのCLASI活動性スコアの変化[治験薬投与後16週時点のスコアが存在しない場合は、last observation carried forward(LOCF)法により利用可能なスコアで補完]
主な副次評価項目
- ベースライン時と治験薬投与後52週時点でのCLASI活動性スコアの変化
- ベースライン時と治験薬投与後16週時点での以下の項目の変化:医師による全般改善度評価(皮膚および皮膚以外)、患者による全般改善度評価(皮膚)、患者の皮膚病変に関わる生活の質(QOL)(Skindex29)、皮膚病変の中央判定、RAPID3[日常生活活動度、原疾患による筋肉または関節の痛み(VAS)、患者による皮膚病変以外の全般評価(VAS)]、倦怠感VAS、BILAG(一般全身症状、筋骨格系症状)および免疫学的パラメータ[抗dsDNA抗体、補体(C3、C4)]
その他の評価項目
- ステロイド減量(治験薬投与後16週以降55週まで)
〈安全性〉
- 有害事象、全ての死亡、重篤な有害事象、治験薬投与中止に至った有害事象、特に注目すべき有害事象、臨床検査値、バイタルサイン、12誘導心電図
- 眼科所見(視力検査、細隙灯顕微鏡検査、眼底検査、視野テスト、色覚検査)
QOL:Quality of Life
RAPID3:Routine Assessment of Patient Index Data 3
VAS:Visual Analogue Scale
BILAG:British Isles Lupus Assessment Group
解析計画:
〈有効性〉
最大の解析対象集団(FAS)を解析対象とし、有意水準を5%、信頼水準を95%とした。CLASI 活動性スコアのベースライン(Day1)と投与後16週時点を対応のあるt検定で比較した。スコアが存在しない場合は、last observation carried forward(LOCF)法により利用可能なスコアで補完した。RAPID3、倦怠感VAS、BILAG 、Skindex29はベースライン(Day1)と投与後16週時点を対応のあるt検定で比較し、医師による全般改善度評価、患者による全般改善度評価、皮膚病変の中央判定、免疫学的パラメータおよびステロイドの投与量は投与群ごとに要約した。
〈安全性〉
安全性解析対象集団を解析対象として、投与期間の有害事象を投与群ごとに要約した。臨床検査値およびバイタルサインについて臨床的に重要な異常を投与群ごとに要約した。
有効性の評価項目
- CLASI: エリテマトーデスの特異疹を活動性病変(活動性スコア)と慢性病変(慢性病変スコア)に分けて評価し、病勢のモニタリングと治療効果を判定する。
- BILAG: 治療の適応と強度を規定する疾患活動性を評価する指標で、1ヵ月間の疾患活動性の動的変化(改善、不変、悪化、新規出現)をとらえ、さらに、疾患活動性を表す症候や検査値異常を各臓器系に分け、半定量化する。
- RAPID3: 筋骨格系症状についての質問などで構成された健康状態に関する患者自己記入式の調査票MDHAQの3つの質問項目「日常生活活動度」、「原疾患による筋肉または関節の痛み(VAS)」および「患者による皮膚病変以外の全般評価(VAS)」の点数を合計しリウマチ性疾患での疾患活動性を算出する。
- 倦怠感VAS: 水平な100mmの直線の左端に「だるさはない」(0点)、右端に「これ以上考えられないだるさ」(10点)と記して、患者に現在感じているだるさの程度の位置にマークをつけてもらい、左端からマークまでの距離を測り(0≦VAS≦100;単位mm)、倦怠感を評価する。
- Skindex29: アンケート用紙の「感情」、「症状」、「機能」の3つの下位尺度に分類される30の質問項目について、5段階(1点:全くなかった、2点:ほとんどなかった、3点:ときどきあった、4点:しばしばあった、5点:いつもそうだった)で患者が回答する。
患者背景(疾患の特性)6)
LEの皮膚病変に基づいた病型の分布および慢性円板状エリテマトーデス(CDLE)の割合は、プラケニル/プラケニル群とプラセボ/プラケニル群で類似しており両群間に大きな偏りはなかった。また、SLE合併の有無別の割合についても両群間に大きな偏りはなかった。
【LE皮膚病変の詳細】
全症例のLEの皮膚病変に基づいた病型別の割合(重複例あり)は、慢性皮膚エリテマトーデス(CCLE)が85.4%と最も高く、次いで急性皮膚エリテマトーデス(ACLE)が18.8%、亜急性皮膚エリテマトーデス(SCLE)が8.3%であった。
CCLEの皮疹の型による分類(重複例あり)では、CDLEが84.4%と最も高く、次いで凍瘡性エリテマトーデス(Chilblain LE)が9.4%、深在性エリテマトーデス(LE profundus)が8.3%、肥厚性エリテマトーデス(Hypertrophic LE)が2.1%、腫脹性エリテマトーデス(LE tumidus)が2.1%であった。
【SLE合併の有無】
米国リウマチ学会のSLE改訂分類基準(1997年)により診断されたSLEの合併の有無別の割合は、SLE合併CLEが56.3%、SLE非合併CLEが43.8%であり、FASの半数以上はSLEを合併したCLE患者であった。
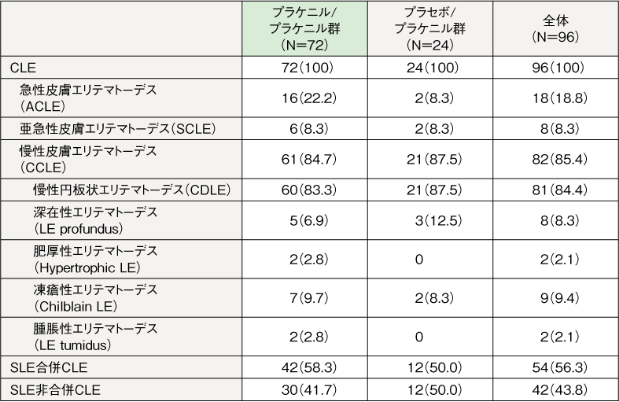
例数(%)
CLEの小分類は重複例あり
本結果は参照群とプラケニル群との比較を示すものではありません。
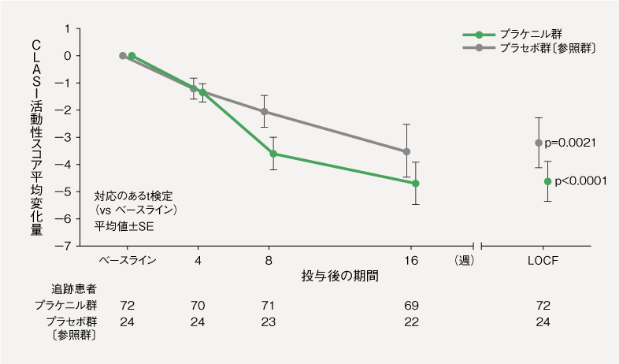
LEの皮膚病変に対する有効性(二重盲検期:投与16週間)6)[主要評価項目]
【CLASI活動性スコア】
主要評価項目である「ベースライン時と治験薬投与後16週時点でのCLASI活動性スコアの変化」について、プラケニル群におけるCLASI活動性スコア(平均値±SD)は、ベースラインで13.5±8.0、投与後16週時点(LOCF)で8.9±6.0、ベースラインからの変化量は-4.6±6.4であり、統計学的に有意な減少がみられた(p<0.0001、対応のあるt検定)。
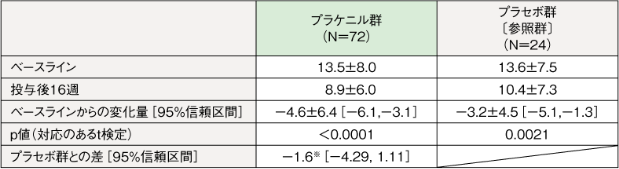
平均値±SD
:本試験では、プラケニル群のプラセボ群に対する優越性を検証するための検出力は考慮されていません。本結果は探索的結果です。
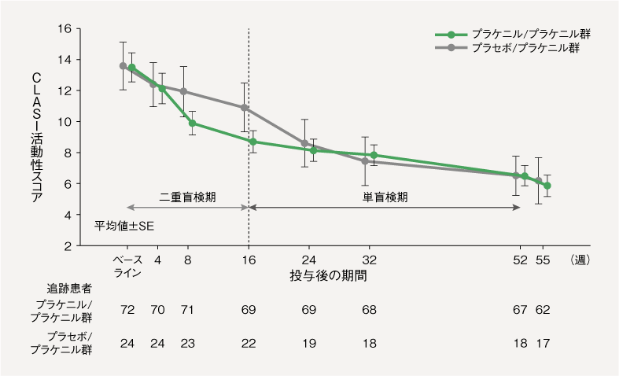
LEの皮膚病変に対する長期有効性(投与後52週まで6)[副次評価項目]
【CLASI活動性スコア】
プラケニル/プラケニル群において、CLASI活動性スコアは、以下のとおり推移した。
その他の皮膚病変の評価6)[副次評価項目]
【皮膚病変に関する医師による全般改善度評価】
治験薬投与後16週時点(LOCF)の皮膚病変に関する医師による全般改善度の「改善」以上(「著明改善」または「改善」)の割合は、プラケニル群51.4%(36/70例)、プラセボ群8.7%(2/23例)、「少し改善」以上(「著明改善」、「改善」、または「少し改善」)の割合は、プラケニル群78.6%(55/70例)、プラセボ群56.5%(13/23例)であった。
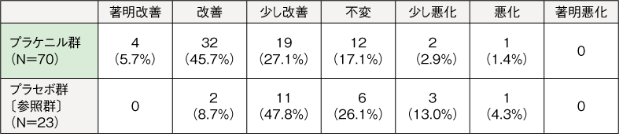
例数(%)
【皮膚病変に関する患者による全般改善度評価】
治験薬投与後16週時点(LOCF)の皮膚病変に関する患者による全般改善度の「改善」以上(「著明改善」または「改善」)の割合は、プラケニル群21.4%(15/70例)、プラセボ群13.0%(3/23例)、「少し改善」以上(「著明改善」、「改善」、または「少し改善」)の割合は、プラケニル群72.9%(51/70例)、プラセボ群47.8%(11/23例)であった。
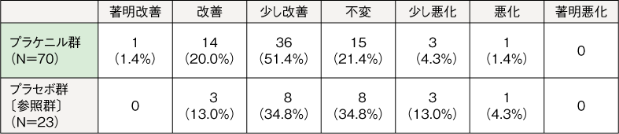
例数(%)
【皮膚病変の中央判定(皮膚病変評価委員による判定)】
治験薬投与後16週時点(LOCF)の皮膚病変評価委員による写真中央判定の「改善」以上(「著明改善」または「改善」)の割合は、プラケニル群59.4%(41/69例)、プラセボ群30.4%(7/23例)であった。
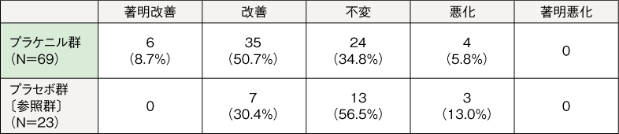
例数(%)
SLEの一般全身症状および筋骨格系症状に対する有効性
(二重盲検期:投与16週間)6)[副次評価項目]
【RAPID3】
RAPID3合計スコア(平均値±SD)は、プラケニル群(N=42)において、治験薬投与後16週時点(LOCF)のベースラインからの変化量は-1.67±3.94であり、統計学的に有意な減少が示された(p=0.0088、対応のあるt検定)。プラセボ群(N=12)においては、ベースラインからの変化量は0.18±4.51であった。
また、プラケニル群における下位項目別でのスコアのベースラインからの変化量(平均値)は、「日常生活活動度」0.05、「原疾患による筋肉または関節の痛み(VAS)」-1.02、「患者による皮膚病変以外の全般評価(VAS)」-0.70であり、「原疾患による筋肉または関節の痛み(VAS)」と「患者による皮膚病変以外の全般評価(VAS)」の両VASで統計学的に有意な減少が示された(日常生活活動度;p=0.6445、筋肉または関節の痛み;p=0.0048、患者による皮膚病変以外の全般評価;p=0.0291、対応のあるt検定)。
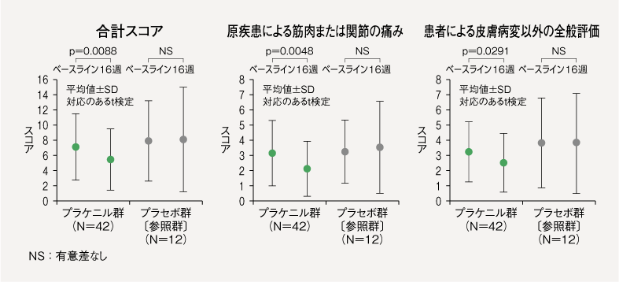
【倦怠感VAS】
倦怠感VAS(平均値±SD)は、プラケニル群(N=42)において、治験薬投与後16週時点(LOCF)のベースラインからの変化量は-1.11±2.48であり、統計学的に有意な減少が示された(p=0.0060、対応のあるt検定)。プラセボ群(N=12)においては、ベースラインからの変化量は-0.71±3.14であった。
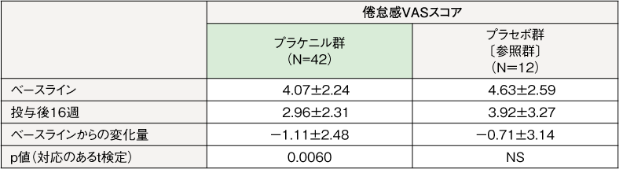
平均値±SD NS:有意差なし
【BILAG(一般全身症状、筋骨格系症状)】
「一般全身症状」と「筋骨格系症状」の両症状群コンポーネントについて、投与前後のスコアを比較した結果、統計学的に有意な減少が示された(一般全身症状;p=0.0331、筋骨格系症状;p<0.0001、対応のあるt検定)。
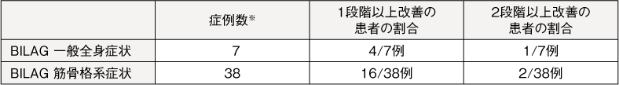
:ベースライン時のカテゴリーがA-Cに該当した患者
【免疫学的パラメータ(抗dsDNA抗体、補体C3、補体C4)】
抗dsDNA抗体価(平均値±SD)は、プラケニル群において、ベースラインで24.39±40.22、治験薬投与後16週時点で21.63±36.81であった。

平均値±SD
補体C3濃度(平均値±SD)は、プラケニル群において、べースラインで76.1±23.7、治験薬投与後16週時点で79.4±21.3であった。

平均値±SD
補体C4濃度(平均値±SD)は、プラケニル群において、べースラインで13.7±6.6、治験薬投与後16週時点で14.9±6.1であった。

平均値±SD
OC(observed care):欠測を補完せず、実際に得られている値で解析
ステロイド減量(治験薬投与後16週以降55週まで)※6)[その他の評価項目]
ステロイド減量の評価では、プラケニル/プラケニル群(FAS)において、ベースライン時にLE治療を目的とした経口ステロイド剤の併用があった患者31例のうち、治験薬投与後に有害事象の対処のため経口ステロイド剤の1日用量が増量された1例を除く30例を対象とした。SLE合併の有無別の内訳は、SLE合併CLEが28例、SLE非合併CLEが2例であり、評価対象患者の多くはSLE合併患者であった。
また、ステロイド増量の評価では、プラケニル/プラケニル群(FAS)において、ベースライン時に経口ステロイド剤の併用がなかった患者41例のうち、投与後に有害事象の対処のため経口ステロイド剤を新規に投与された1例を除く40例を対象とした。SLE合併の有無別の内訳は、SLE合併CLEが12例、SLE非合併CLEが28例であった。
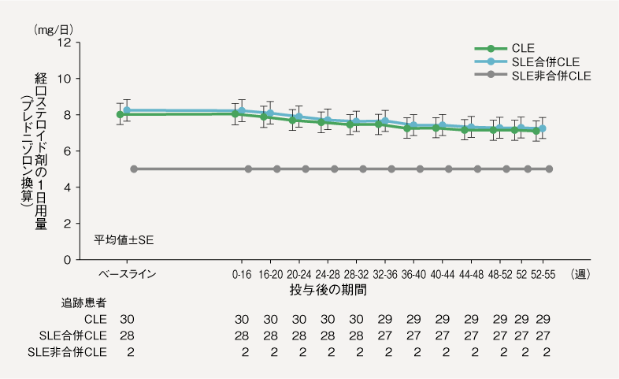
プラケニル/プラケニル群において、LE治療を目的とした経口ステロイド剤のプレドニゾロン換算による1日用量(時点または期間別集計による1日当たりの平均値)は、ベースラインで8.01mg/日(中央値は8.00mg/日)に対して、投与後52週来院時で7.08mg/日(中央値は6.00mg/日)であった。
また、ベースライン時にLE治療を目的とした経口ステロイド剤の併用があった患者では、投与後52週来院時に31.0%(9/29例)で経口ステロイド剤の1日用量が減量され、69.0%(20/29例)でベースライン時の1日用量が維持されており、52週間の投与期間を通じてベースラインの1日用量を超える増量が行われた患者はいなかった。
LE治療を目的とした経口ステロイド剤の1日用量(プレドニゾロン換算)の推移(プラケニル/プラケニル群)- FAS
参考情報6)
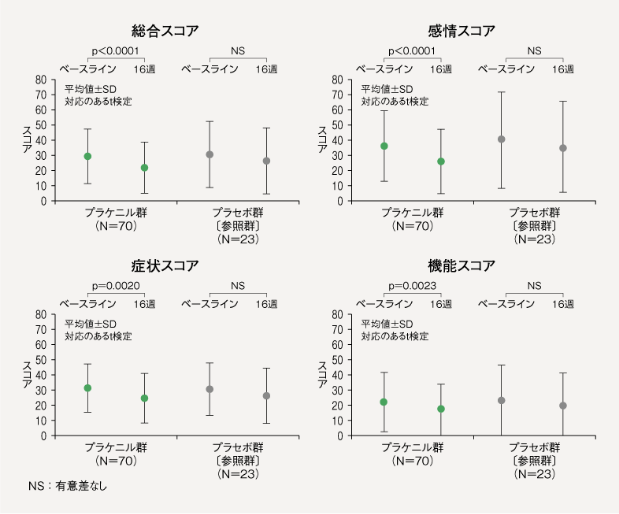
【皮膚病変に関わるQOL(Skindex29)】[副次評価項目]
Skindex29総合スコア(平均値±SD)は、プラケニル群(N=70)において、治験薬投与後16週時点(LOCF)のベースラインからの変化量は-7.43±14.44であり、統計学的に有意な減少が示された(p<0.0001、対応のあるt検定)。プラセボ群(N=23)においては、ベースラインからの変化量は-4.30±13.80であった。
また、プラケニル群における下位尺度ごとのスコアのベースラインからの変化量(平均値)は、「感情」-10.18、「症状」-6.88、「機能」-5.51であり、全ての下位尺度でスコアの統計学的に有意な減少が示された(感情;p<0.0001、症状;p=0.0020、機能;p=0.0023、対応のあるt検定)。
6)承認時評価資料(国内第Ⅲ相試験)
製品に関するご不明な点は、
くすり相談窓口までお問い合わせください。
くすり相談窓口
受付時間:9:00〜17:45
(土日祝、休業日を除く)
当社は、日本製薬工業協会が提唱する
くすり相談窓口の役割・使命
に則り、
くすりの適正使用情報をご提供しています。